C-C Bond Cyclic Peptide Formation
C-C Single Bond Formation
Traditional Cross Coupling
Cross coupling corm the new carbon-carbon bond by transition metal catalysis. In most reactions, the transition metal like Pd inserts into a carbon-halide bond, then the subsequent trans-metallation of an organo-metallic compound induces two organic fragments bound to the catalyst. The new carbon-carbon bond will form by reductive elimination. Most typical C-C cross couplings are used to synthesize peptide macrocycles. But this is less often than copper-catalyzed azide-alkyne cycloaddition (CuAAC).
CH-Activation Coupling
CH-activation coupling, in contract to traditional cross coupling, do not require the introduction of organ-metals in the substrates. This is a critical advantage, since strongly basic groups are problematic for peptide formations. In these reactions, the CH-group reacts with the organic halide as a nucleophile.
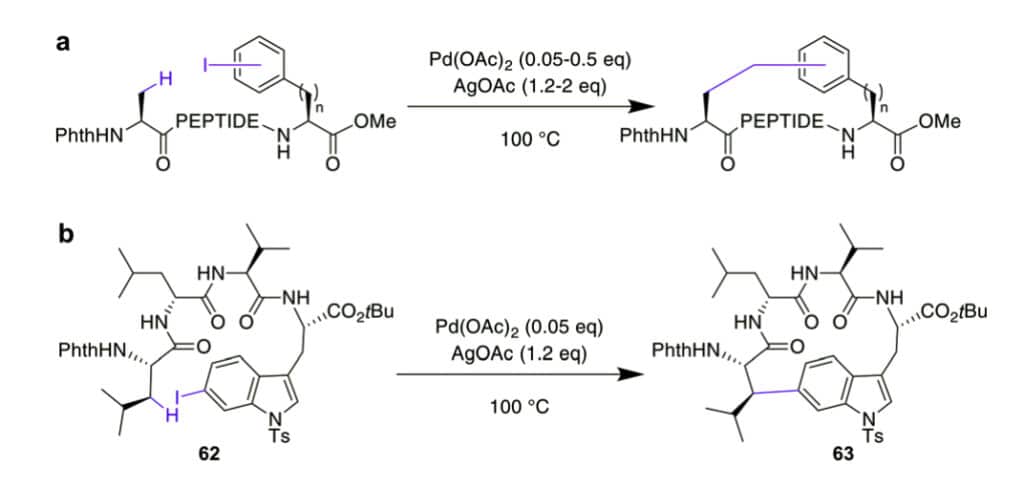
CH activation can modify the C2 of indole in Trp selectively, these reactions can couple aryl halides to the assembled peptide by Pd-catalysis.
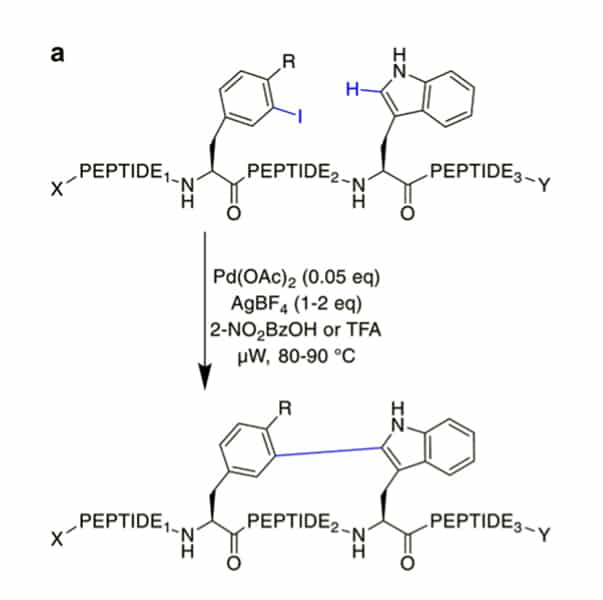
The cyclic peptides are stable against proteolytic degradation, and most amino acids are tolerated. Once acylated with picolinic acid, this reaction can couple phenyl iodides with sp2-CH groups at the γ or δ position in the N-terminus.
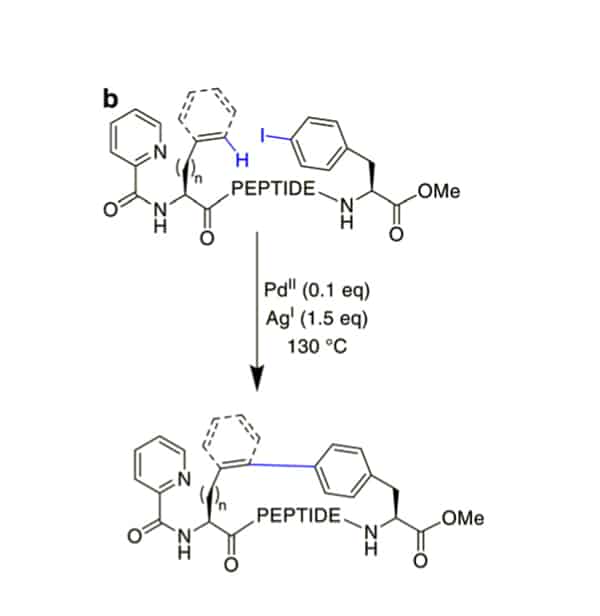
In order to remove the introduction of aryl halide, the phenyl residues can couple to terminal alkenes under Pd-catalysis with added AgOAc.
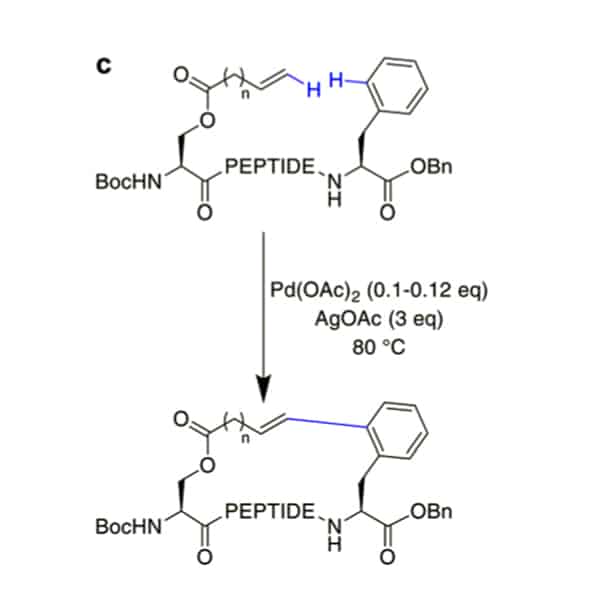
Phthaloyl-protection of N-terminus in peptides result in modified acidity, this will activate the β-hydrogens on aliphatic sidechains specifically, then couple with aromatic halides for cyclization in Pd-catalysis.
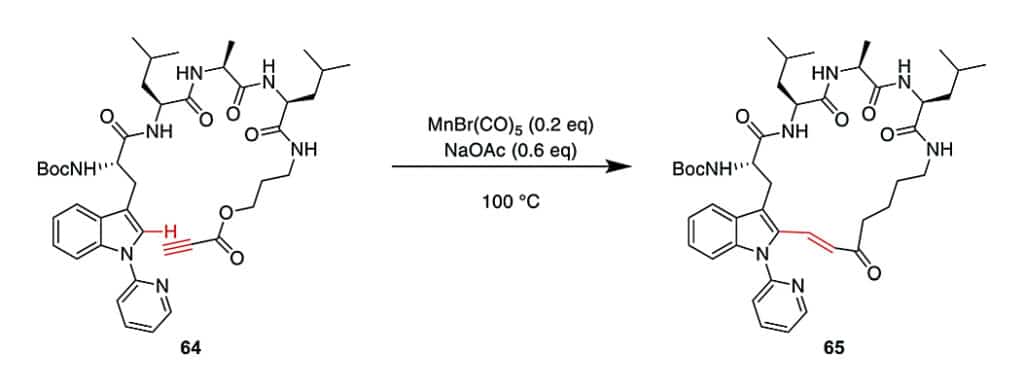
Except Pd, Mn is a new transition metal for CH activation, it can alkylate the indole-C2 of N-pyridine Trp, where pyridine is essential as a directing group for selectivity. Combining this CH-activation with the propargylic ester, in order to generate α, β-unsaturated esters. Which are versatile handles for further derivation of cyclo- or conjugate additions.
Photocatalyzed Reactions
Unfunctionalized iridium (Ir) photoredox reactions can lead to peptide macrocyclization. Under light irradiation, the Ir-catalyst will promote radical formation on C-terminal carboxylate, and lead to decarboxylation. The remaining carboxylate proceeds 1,4-addition with acrylates or malonates.
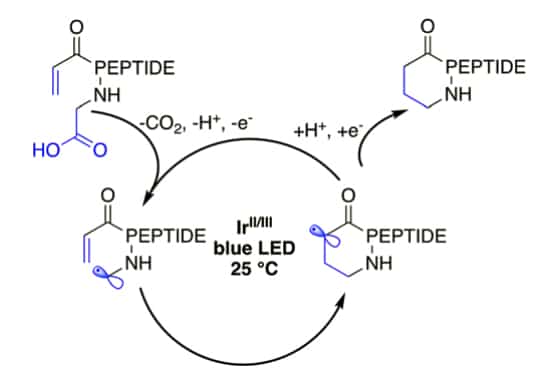
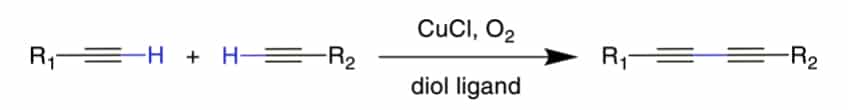
Glaser–Hay Coupling
Glaser-Hay reaction can couple two alkynes to a dialkyne, and create a very rigid and extended bridging group. The Glaser-Hay coupling occurs in O2 presence with Cu-catalysis. The resulting bis-alkyne can be reduced in catalytic hydrogenation process.
Alkene Metathesis(C-C Double Bond)
Alkene metathesis can form two new alkenes between two alkenes in the chemoselective and stable catalysts of molybdenum (Mo)- and ruthenium (Ru)-based. This reaction is applied prominently for cyclic system generations, and ring-closing metathesis (RCM) labeling. Alkene metathesis has high tolerance to functional groups, and high yields. This makes it suitable for cyclizaton of relatively large and functionally diverse molecules. Therefore, this reaction method is employed in large number of complex structures, like peptides and peptidomimetics.
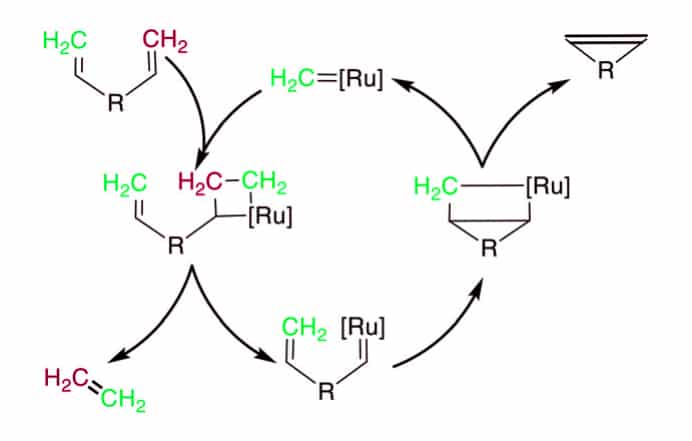
The Ru catalyzed reaction mechanism of ring-closing metathesis(RCM):
- The carbene or alkylidenne with catalyst-bond react with alkene to form a [2 + 2]-cyclization, and generate the metalla-cyclobutane intermediate.
- The subsequent ring opening will bind the catalyst to the substrate, this results in the release of another akene (ethene as a terminal alkene substrate).
- The second [2 + 2] cycloaddition occur with the second alkene on the substrate.
- The final release will open the metallacyclobutane to create the bridged product and catalyst.
This cyclic reaction is applied to peptide for stabilizing secondary structures, especially for α-helices. Moreover, it is also used to stabilize or mimic other motifs, like β-sheets, β-turns, polyproline II helices, N-capping boxes, disulfide bridges.
Alkenes for peptide stapling are introduced as modified side-chains on carbon, α-N, side-chain aliphatic alcohols and phenols, C-terminal or side-chain acids, N-terminal carbamates, and cysteine thiol groups. There is an RCM formation of 15-membered macrocycle to produce an anti-hepatitis C peptidomimetic.
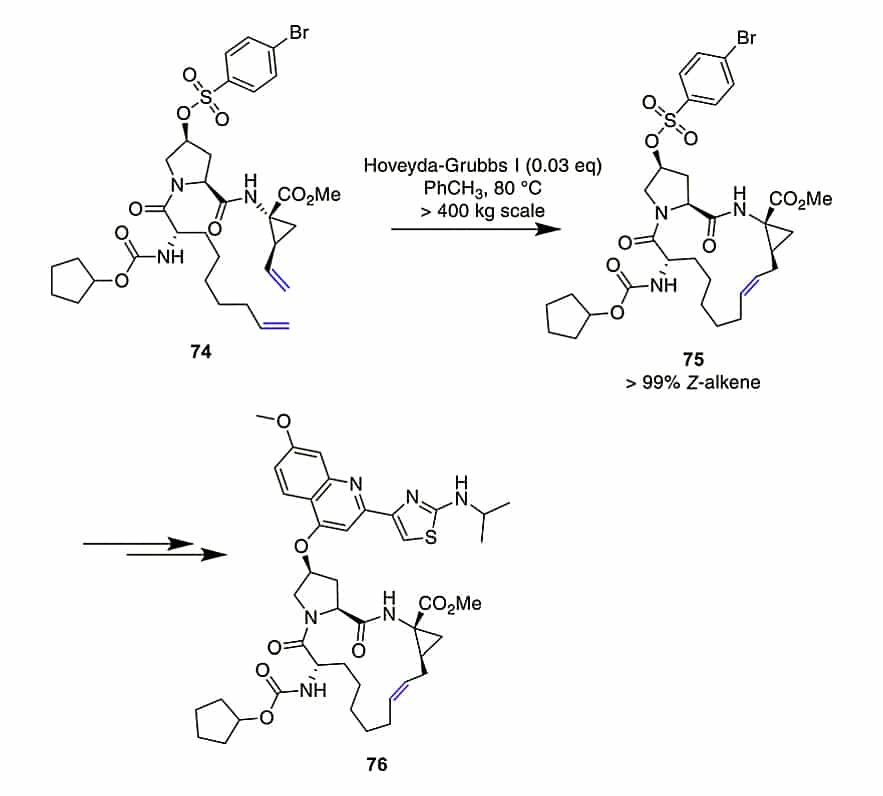
The ring-closing metathesis (RCM) demonstrate flexibility, it can be applied to unprotected oxytocin, octreotate, two α-conotoxins, and an insulin fragment. The selection of solvent is crucial, the addition of chaotropic agent(guanidinium·HCl) in neutral conditions will improve the yield substantially.
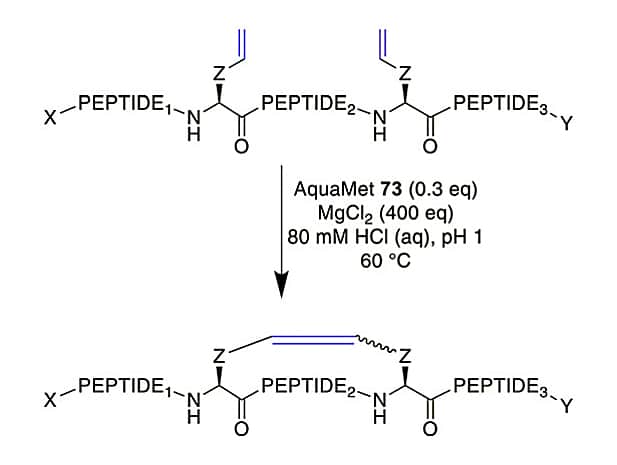
Furthermore, RCM is also employed to optimize DNA-encoded libraries. Improved acidic conditions will mask coordinating groups, in order to achieve α-helical stapled peptide with 52-65% conversion, this depends on the linker length between peptide and DNA tag.
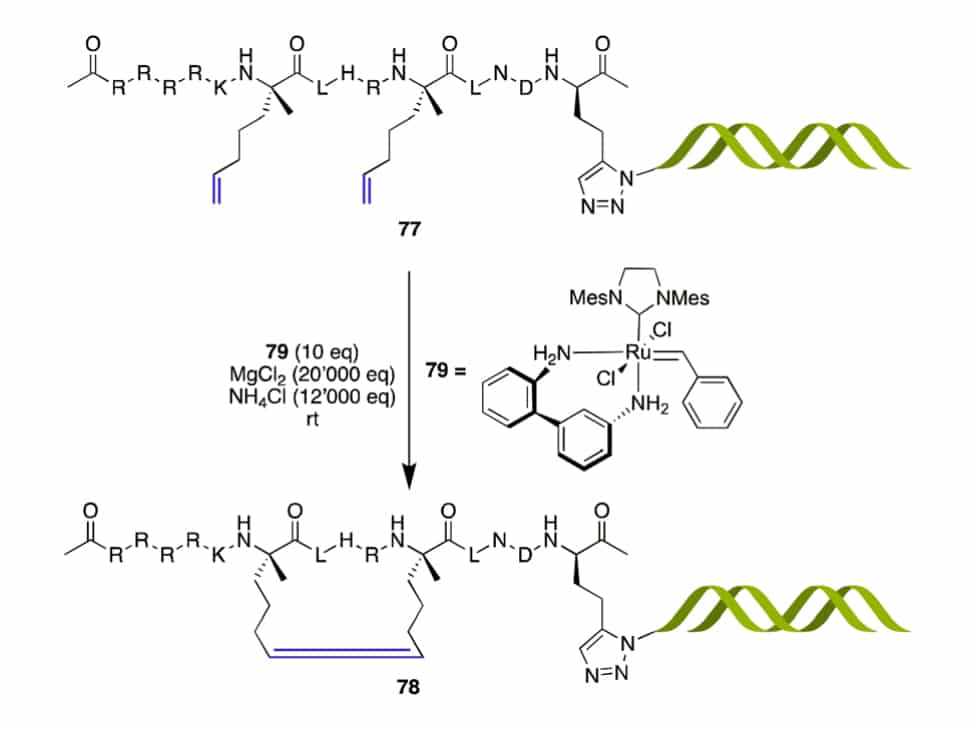
Ring-Closing Alkyne Metathesis(RCAM)
The ring-closing alkyne metathesis (RCAM) with tungsten (W) or molybdenum (Mo) catalysis generates a new alkyne in the similar way to alkene metathesis. However, the RCAM results in extended conformation and fewer isomers. The common catalysts are high-valency W or Mo basic compounds.
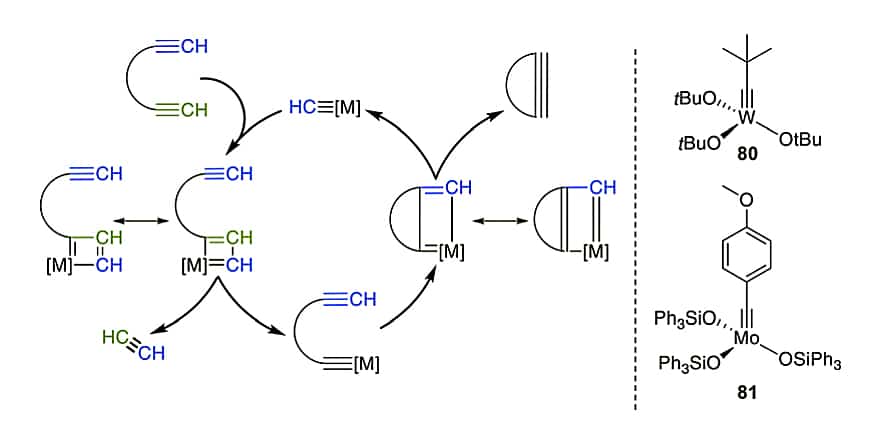
The reaction mechanism of ring-closing alkyne metathesis(RCAM):
- The catalyst-bound alkylidene cyclo-add with the alkyne to form a metalla-cyclobutadiene.
- the compound ring open, the substrate bind to the catalyst.
- Cyclo-addition with another alkyne to form a new metallacycle.
- After another ring opening, generate the final cyclic product and catalyst.
Despite the similarities, RCAM has less application to peptides than RCM. Nicely, the RCM and RCAM can be performed selectively with alkynes and alkenes and in possible orders.