Chemical Synthesis of Unnatural Amino Acids
Introduction
Unnatural amino acids (UAAs) not exist in natural polypeptide chains, so they are considered non-proteinogenic amino acids. They are an important component in the manufacture of many drugs and expand the range of protein functions via integrating them into proteins. Based on its structure may be similar or significantly different from natural amino acids, UAA can be divided into two categories, which are usually called analogues or substitutes, respectively. Regardless of which class they belong to, UAAs are highly attractive as bioactive molecules or building blocks owing to unique chemical and biological properties derived from their unusual side groups and/or d-stereochemistry.
The synthesis and applications of UAA has attracted a lot of interest, especially from the enzymology and drug development domains. UAA has the ability to introduce altered physicochemical and biological properties by replacing any natural amino acid at a certain point. This capacity has become a valuable molecular tool in protein project. It can be added to a variety of structural elements to develop potential leads in complexes that are both peptidic and non-peptidic. The insertion of UAAs into proteins widens their functional diversity, including optical probes, protein labelling, signal transductionand post-translational modifications, etc. In addition, UAAs significantly increase the potential applications of chiral building blocks and molecular scaffolds in the construction of combinatorial libraries.
Chemical Synthetic Technology
There have been considerable efforts to develop various synthetic ways to obtain structures based on amino acids. The methods for UAA synthesis mainly have two types: biosynthesis and chemical synthesis. Compared with the biocatalysis route, Chemical routes are more profound, which is attributed to the diversity of synthesis methods. Many chemical synthesis methods can be used to prepare UAA.
Asymmetric Synthesis
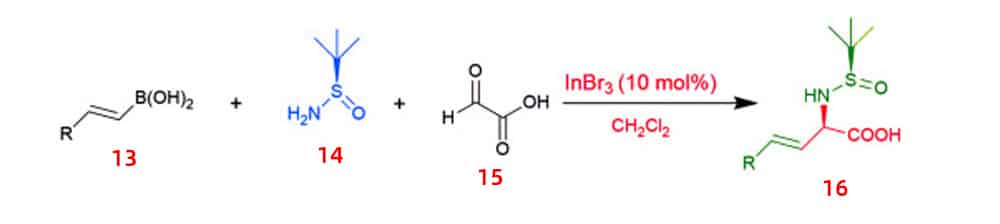
Asymmetric catalytic synthesis is the most efficient and effective way in the production of various optically enriched α-amino acids, and is widely used in the chemical synthesis of pharmaceuticals. The asymmetric synthesis of β,γ-unsaturated α-amino acids 16 is accomplished mainly by Lewis acid-promoted highly diastereoselective Petasis three-component-reaction of vinylboronic acid 13, N-tert-butylsulfenamide14 and glyoxylic acid 15. In order to maximize yield and diastereoselectivity, this reaction required to be carried out at room temperature.
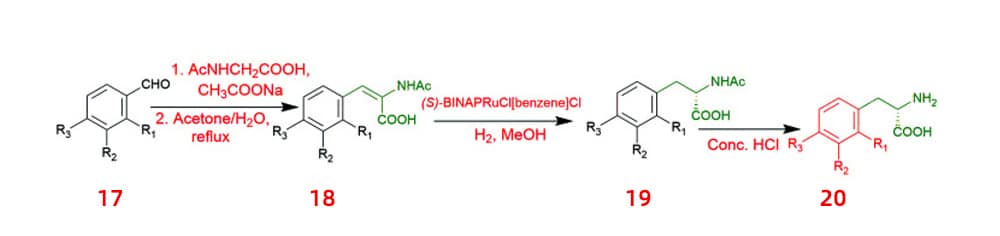
Catalytic methods showing high levels of enantioselectivity control have attracted special attention from scientists. N-acetylamino phenyl acrylic acids 18 were hydrogenated asymmetrically with a ruthenium catalyst to produce chiral acids 20, which are anticipated to have significant applications in medicines.
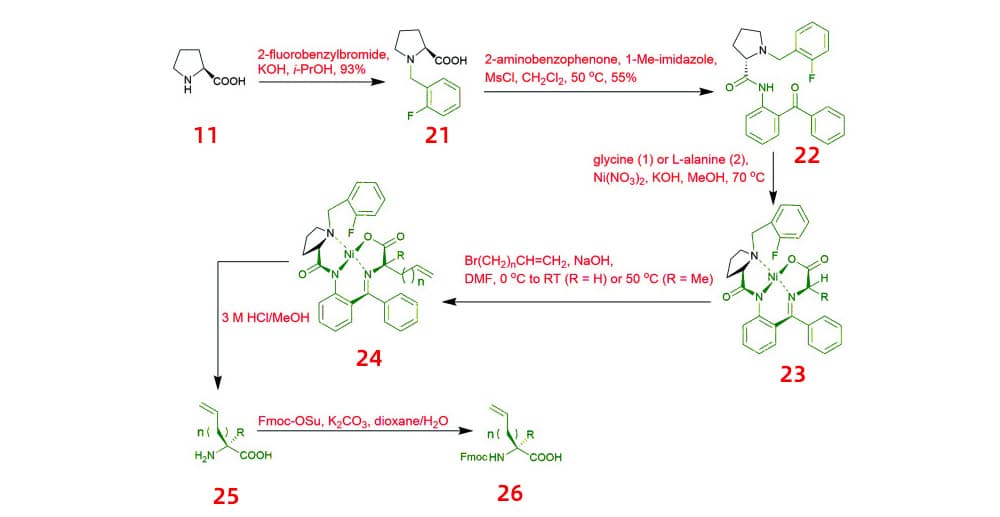
Bioactive conformation is facilitated via conformationally constrained peptides because of their lower folding penalty. Conformationally constrained peptides have given rise to a new class of chemical and pharmacological development tools. An unnatural alkenyl amino acids (25, 26), which is required for peptide “staple”, are generated by asymmetric alkylation of fluorine-modified Ni(ii) Schiff base complexes (23, 24).
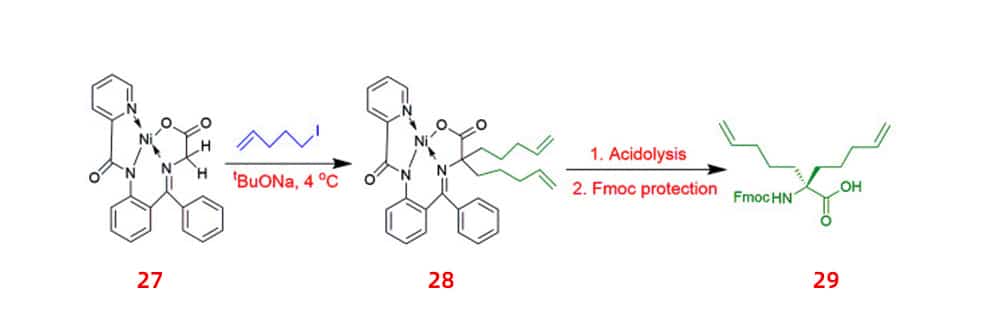
The alkylation of glycine is the most obvious method for making UAAs. Switching to chiral phase transfer catalysts that more economical and environmentally friendly, the yield of asymmetric alkylation of glycine varies from 60% to 90%. A method was developed to minimize impact toxic reagents because these synthetic routes involve toxic reagents. Starting from proline, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/4-Dimethylaminopyridine (EDCI/DMAP) was used as a coupling agent to synthesize the derivative of glycine non-proline 27,a chiral intermediate,which was then alkylated in the presence of sodium tert-butoxide to afford 28. This method produced five UAAs for peptide stapling 29 with yields ranging from 60% to 70% and high enantioselectivity >99% ee.
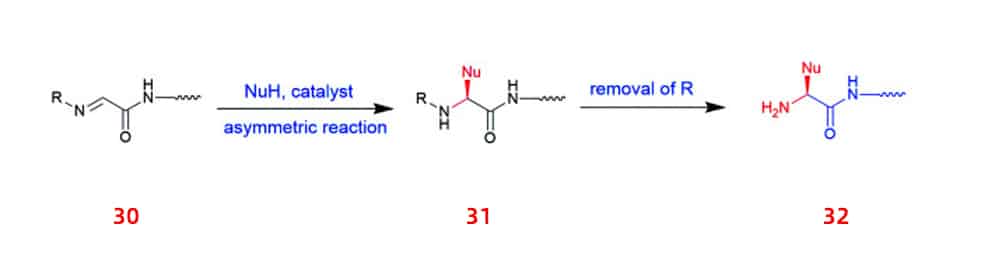
In addition, a new approach using imino peptides 30 (e.g., α-imino perfluro alkylesters, imides, or thioesters) to asymmetrically synthesize UAA-containing peptides 32 was proposed.
Diastereoselective Synthesis
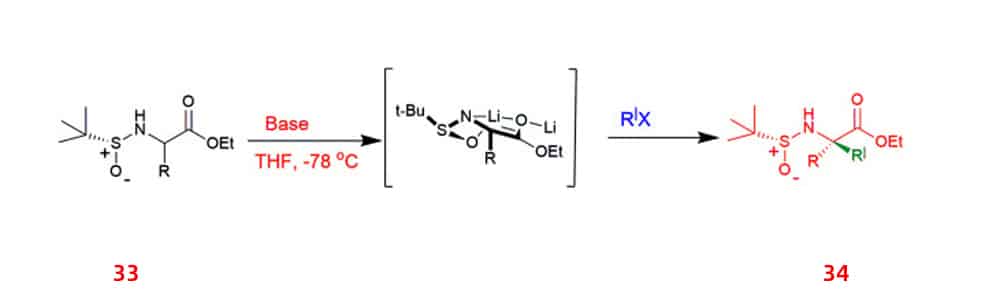
One method for producing UAA is new diastereoselective synthesis, which is also an important area of organic synthesis research. Example that the diastereoselective synthesis of UAA via alkylation of α-tert-butyrasulfinamide 33 auxiliary-bound the enolate, in which a chiral α-sulfenamido ester that is alkylated under the prevailing conditions produced up to 90% of mono and α, α-disubstituted amino acid derivatives 34.
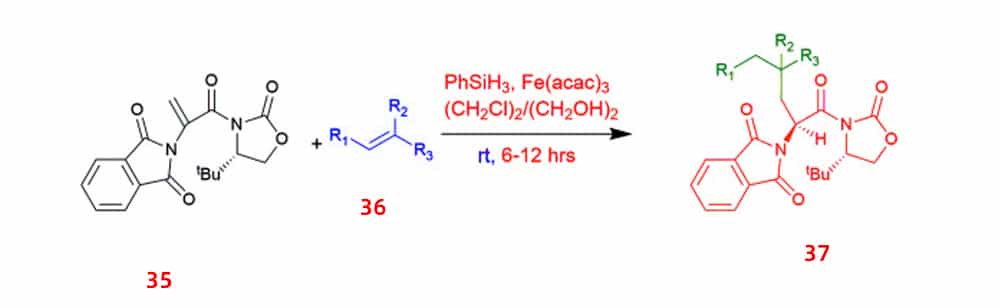
Another approach is iron-catalyzed diastereoselectivity synthesis. This approach yielded in unnatural chiral (S)-α-amino acids with a γ-quaternary carbon center 37 by employing the widely used iron salts, 2-phthaloylacrylamide 35 and alkenes 36 as starting materials, and phenylsilane as reducing agent.
Enantioselective Synthesis
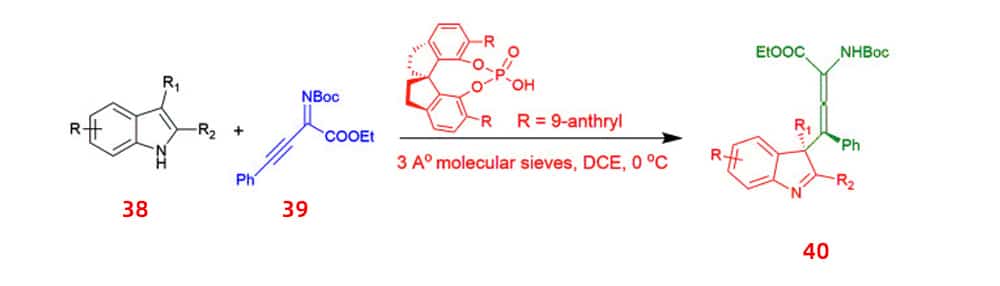
The synthesis of enantioenriched allenic compounds has attracted considerable attention. One of the methods that has been developed is the first asymmetric synthesis of tetrasubstituted α-allenoates 40, which was performed mainly by using chiral phosphoric acid catalyzed dearomative γ-addition reaction of 2,3-disubstituted indoles 38 to β,γ-alkynyl-α-imino esters 39.
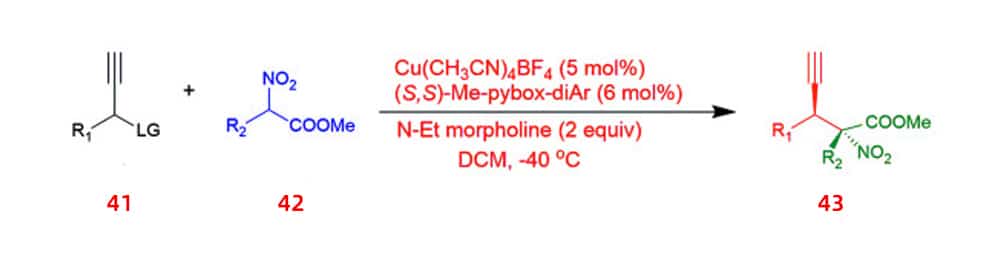
A method is proposed for enantioselective propargyl substitution reactions catalyzed by Cu-pybox complexes. This method resulted in high yields of non-protein quaternary α-amino acid precursors 43 with two stereocenters and a terminal alkyne moiety by using propargyl carbonate 41 and α-substituted nitroacetates 42.
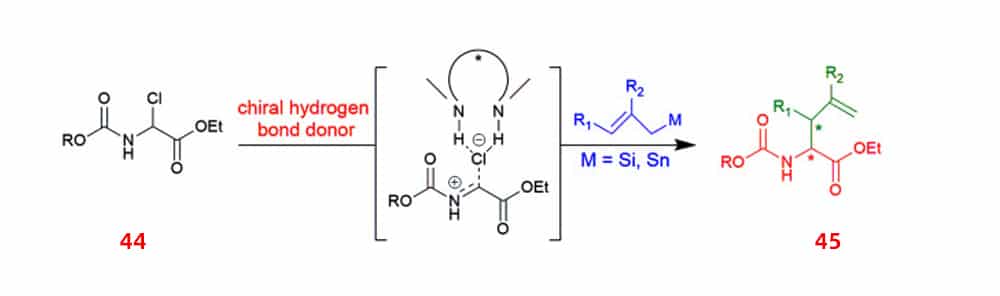
Since its diverse range of applications, the synthesis of α-allyl amino acids has received significant interest. Utilizing chiral squaramide hydrogen bond donor anionic abstract catalysts, α-chloro glycine esters 44 was allylated to produce α-allyl amino esters 45 in a enantioselectivity manner.
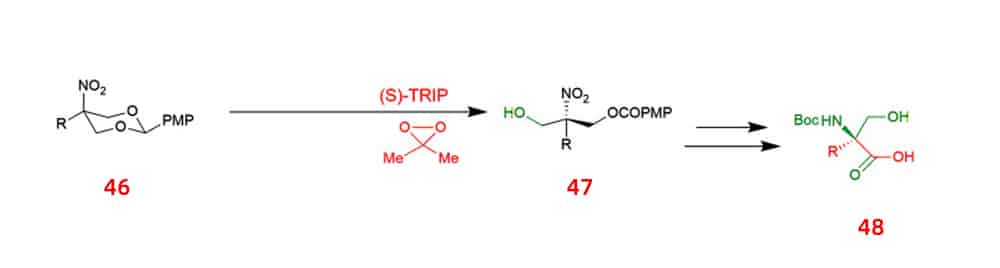
Another approach is to use asymmetric desymmetry to synthesize acyclic α-tertiary amines with high enantioselectivity. This method relies on DMDO as the oxidant, and oxidative desymmetrization of 2-substituted 2-nitro-1,3-diolbenzylidine acetals 46 is mediated by the chiral phosphoric acid. This process generates chiral 2-nitro-1,3-diols with great enantioselectivity that could be converted into optically pure, unnatural α-alkyl serine 48.
Catalytic Synthesis
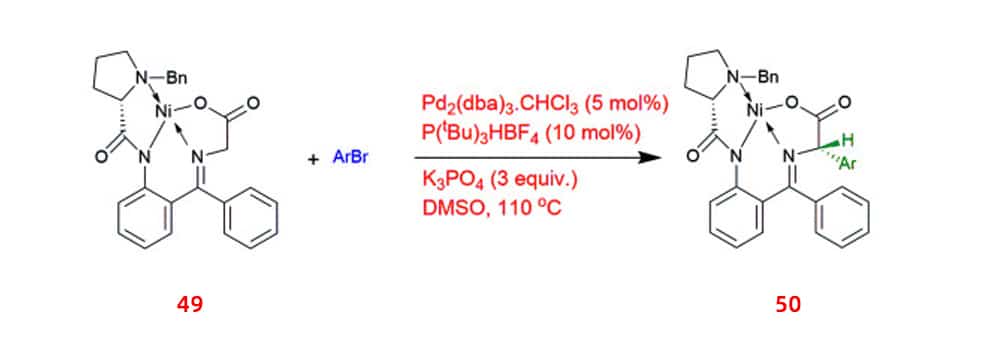
Many bioactive substances and natural products contain substituted arylglycines as structural motifs, which has prompted attempts to synthesis acylglycines. It has been reported that Pd-catalyzed α-arylation of a chiral nickel(II) glycinate complex 50 with different aryl bromides enabled the stereoselective synthesis of optically active arylglycine derivatives 49 in yields as high as 80%.
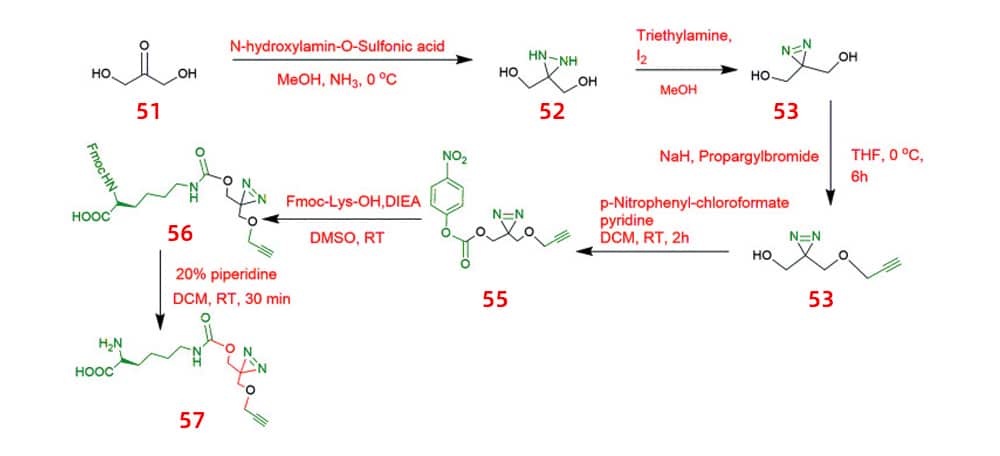
Lysine-based bifunctional amino acids react with azides using copper-catalyzed click chemistry. Dihydroxyacetone 51, which is marketed commercially, served as the starting material. After two steps of reaction, ketone was changed into diazepine (52, 53) in liquid ammonia. One of the two hydroxyl groups is then alkylated by using propargyl bromide. Next, the isolated monosubstituted product 54 is altered into a reactive carbonate and conjugated to lysine (56, 57).
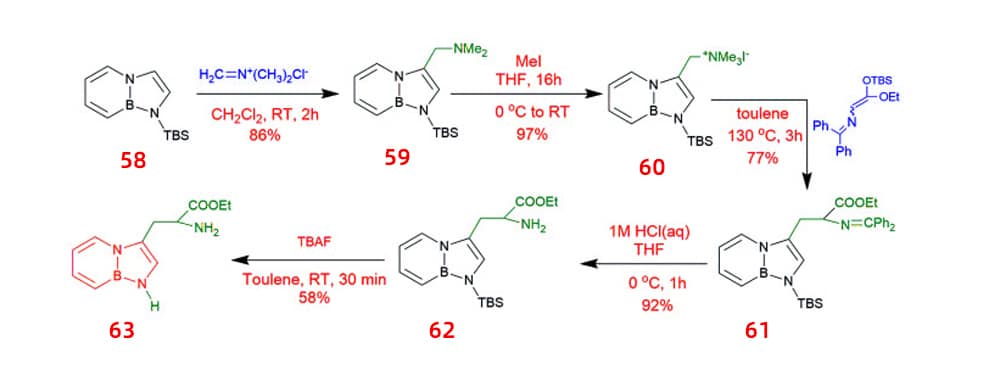
Tryptophan BN analogues combined with UAA may provide a way to examine the fluorescence and functional properties of proteins. Functionalization of BN-indole 58 has also been used to synthesize unnatural analogues of tryptophan 63 containing B and N. Dimethyliminium chloride was used in a substitution process with TBS-BN-indole 58. The resultant product 59 was methylated with iodomethane and subsequently replaced with silyl-ketene-acetal. In an acidic aqueous environment, the Schiff base protective group of 62 can be eliminated. BN-tryptophan ethyl ester 63 was then produced by deprotecting the silyl protecting group.
Chiral Pool Synthesis
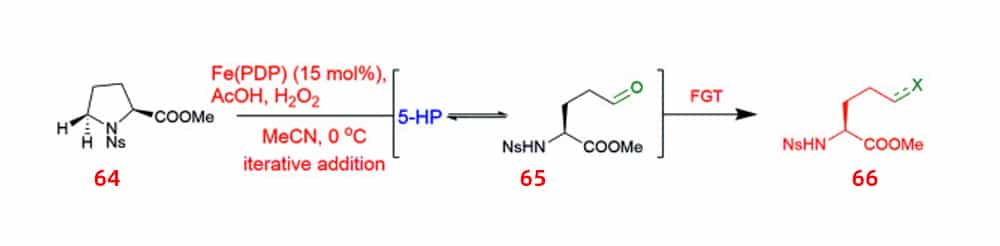
Chiral pool synthesis can be used to broadly improve UAA production. A method for targeted C–H oxidative alteration of amino acids and peptides while maintaining α-center chirality using iron catalysts is proposed. In this method, substituted proline 64 can be oxidized to 5-hydroxyproline, which produces an intermediate 65 with a highly synthetically versatile hemiaminal functional group that can be converted to UAAs and peptides containing UAA 66.
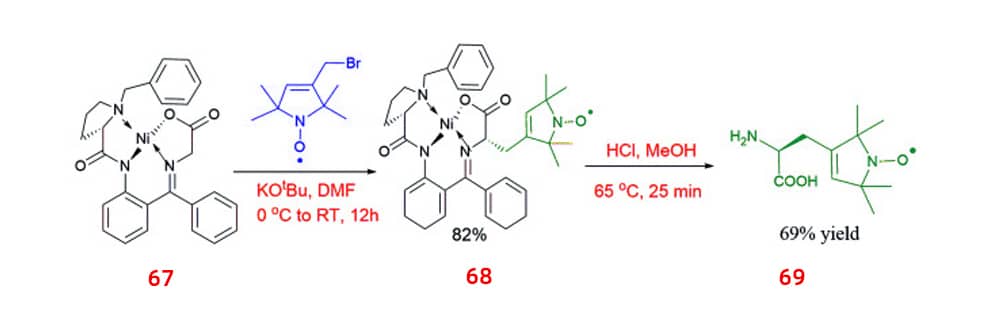
Alternatively, chiral spin-labelled amino acids 69 ,which were produced by cleavage from their respective complexes (Belokon complex) 67, were introduced to d-amino acid oxidase (DAAO) in order to improve enantiopurity. α-keto acids that selectively converted by d-amino acids can be separated from the desired l-amino acids via using this catalytic process.
Conclusion
Currently, UAAs are already used on the market and obviously has a lot of room for growth in their application. For the synthesis of UAA, despite chemical synthesis is not ecologically friendly, it is non-tedious and robust. From now on, a novel synthesis technique for UAA may be to eliminate its toxic effects through a minute changes in chemical processes with green chemistry.