Peptide Macrocyclization Strategies
Introduction
Macro-cyclic peptides are a critical molecular format in drug discovery, these peptides combine advantages of small-molecule and biological therapeutics. Such as: synthetic accessibility, low immunogenicity and toxicity, high binding affinity and selectivity, targeting ability of undruggable protein surfaces. Furthermore, macrocyclization of peptides have higher stability and increased membrane permeability, this is a critical medicinal chemistry strategy in peptide drug development.
The synthesis of cyclic peptides is difficult in traditional methods, like amide formation. Since the defined pre-cyclization conformations must be formed before the desired intramolecular reactions, but this is an entropically unfavorable process. Especially for head-to-tail cyclization, involving the cyclization of the C-terminus with its N-terminus.
Besides the head-to-tail, peptides are also cyclized by head-to-sidechain, sidechain-to-tail, sidechain-to-sidechain. In particular, sidechain-to-sidechain cyclization are applied extensively to stabilize secondary structures, such as α-helices and β-sheets. These peptides are also called as stapled peptides, and generating protein epitopes and antibody CDR mimetics.
There are great progress in new cyclization strategies for peptide macro-cyclization, involving chemistry from cross-coupling and photochmical reactions to enzymatic macrocyclization. In addition, chemo-selective reactions with orthogonality and diversity push macro-cyclic peptides to new and drug-like modalities. In this article, we compile the most critical and modern organic chemistry macrocyclization strategies in peptide synthesis.
Disulfide Cyclization
Since the high nucleophilicity of thiolate, cysteine is the most suitable amino acids for chemical transformation of linear peptides. Disulfides are the common structural motif in peptides, proteins and other natural compounds, in order to stabilize tertiary structures and conformations.
Disulfide is easily formed between two proximal thiolates in oxidative environments. However, disulfides are inherently unstable in the reducing environment. In order to improve the stability, disulfide groups are replaced with selenium, actam, thioether, triazole or dicarba analogues. Most replacement methods require significant modifications of the synthetic building blocks.
Thioacetal Formation
The thioacetal is a bridging motif, it is a flexible and reduction-stable analogues for native disulfide bridges. The S-S distance in the methylene thioacetal is 2.95 angstrom, while 2.02 angstrom in the disulfide.
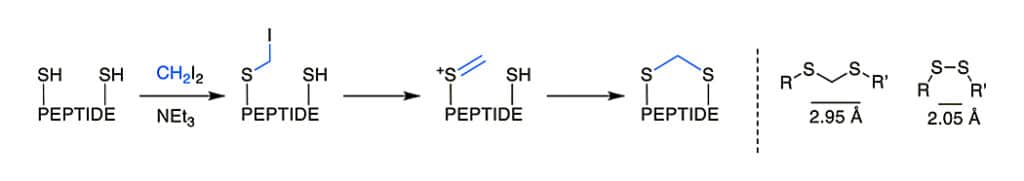
The thioacetal formation is possible by Kourra and Cramer procedure, in aqueous environments and under mild conditions without protecting groups. Once thiols or situ-reduced disulfides react with diiodomethane and a base, they generate the methylene thioacetal with good yield.
Thioether Formation
The nucleophilicity of cysteine thiols can form sidechain-to-sidechain stapling through thiother formation. The bromo or chloroacetate induce α-helicity in peptides as reactive moieties, which couple to sidechain or N-terminus, like ornithine, O-[2-bromoethyl]-tyrosine. The thioether forms in aqueous buffers at pH8.
The thioether bond for cyclization is generated by the thiol-ene reaction on solid support, the radical thiol group add to an alkene (allyloxycarbonyl protecting group). This radical reaction is initiated in the ultraviolet light irradiation and a radical initiator.
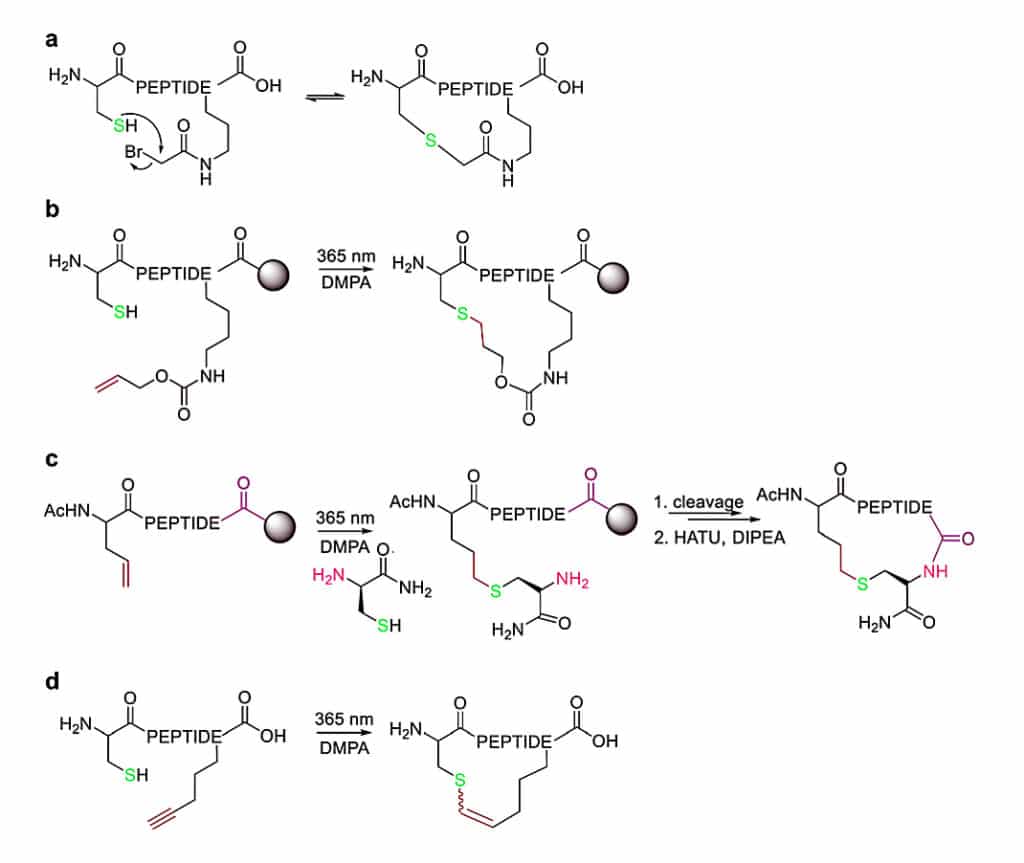
Similarly, the radical thiol-ene reaction can occur between the cysteine thiolate and an alkene-containing noncanonical amino acid. The cyclization is achieved by conventional amide formation between the amine of the N-terminal Cys and the C-terminus.
In addition, thiol-yne coupling is another thioether-cyclization, the alkyne-containing amino acid reacts with cysteine under photo-induction, then yield mixtures of E- and Z-isomers.
Ether Formation
Ethers is an interesting peptide bridging motif, it is flexible with multiple conformations. It is more stable to reduction, oxidation or nucleophiles than disulfide or thioether. However, there are limited applications in peptide macrocyclization. Biaryl ethers has the critical role in peptide synthesis, in particular for antibiotic glycopeptides – vancomycin.
There are several methods for the synthesis of macrocyclic biaryl ether peptides. Generally, all these methods require protective group strategies and custom-made building blocks.
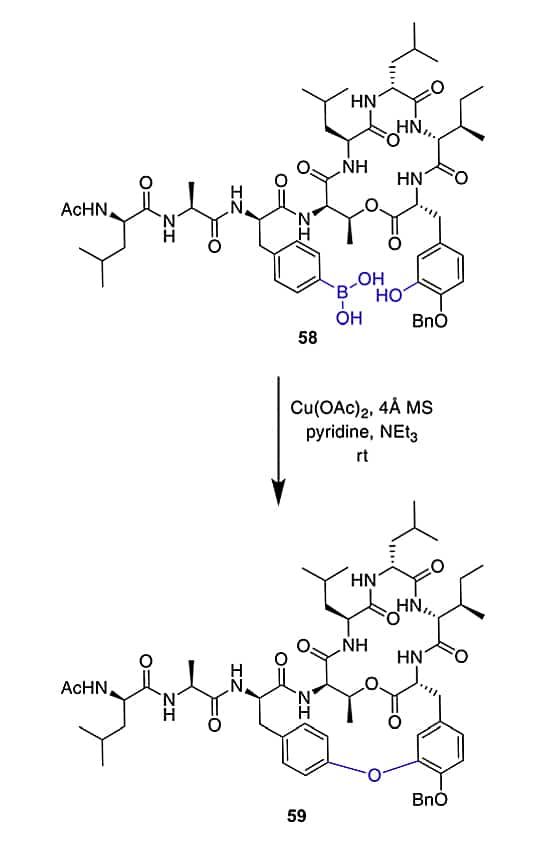
Evans–Chan–Lam Reaction
The phenyl boronic acid is introduced as pinacol ester, this is stable through amide coupling conditions and TFA-mediated cleavage from the solid phase. Then the pinocol ester liberate prior to the Evans-Chan-Lam reaction.
Tsuji–Trost Reaction
Tsuji-Trost reaction applies allylic esters to couple different native sidechains. In conditions without carboxylates, amines, histidines, or cysteines, this reaction is specific for tyrosine as the nucleophile.
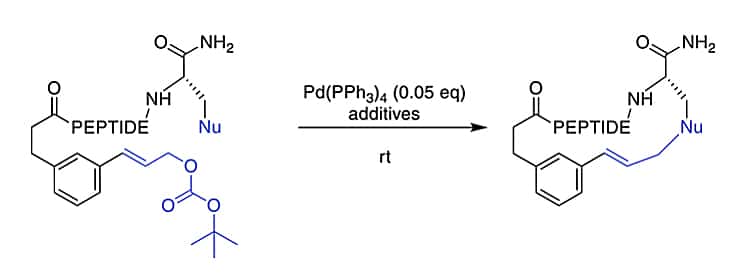
Ni/Photoredox Catalysis
Ni/Photoredox catalysis can enable hard-to-imagine bond formation in peptide macrocyclization. It forms the ether bond between the C-terminal serine and a 2-bromobenzoyl moiety at the N-terminus. However, if the C-terminus is an amide instead of an ester, it will react via the amide nitrogen.
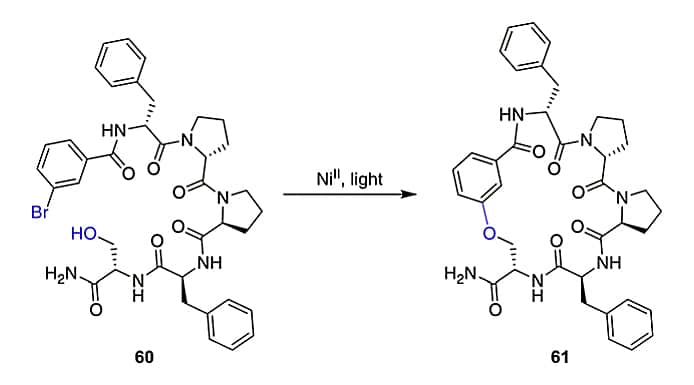
Triazole Formation
1,4-Disubstituted Triazole Formation(CUAAC, Click Chemistry)
The copper-catalyzed azide-alkyne cycloaddition is a Huisgen 1,3-dipolar cycloaddition, it is an exemplary click reaction. The Cu-mediated catalysis generate 1,4-substituted 1,2,3-triazoles from the terminal alkyne and azide.
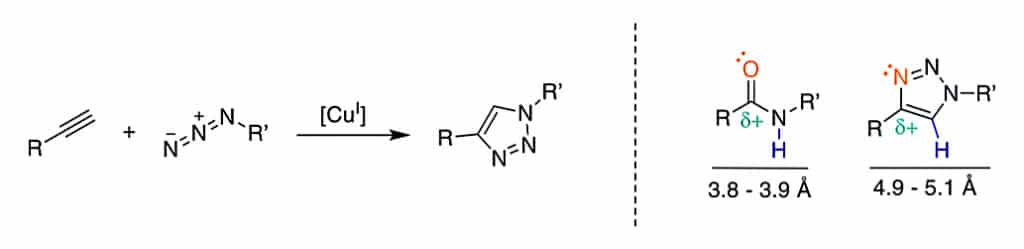
This 1,2,3-triazole is robust, selective and water-compatible. It can mimic the Z-amide in terms of H-bond acceptors and donors. Furthermore, the triazole has better dipolarity than the amide. The 1,2,3-triazole are applied to stabilize the secondary structures or other motifs.
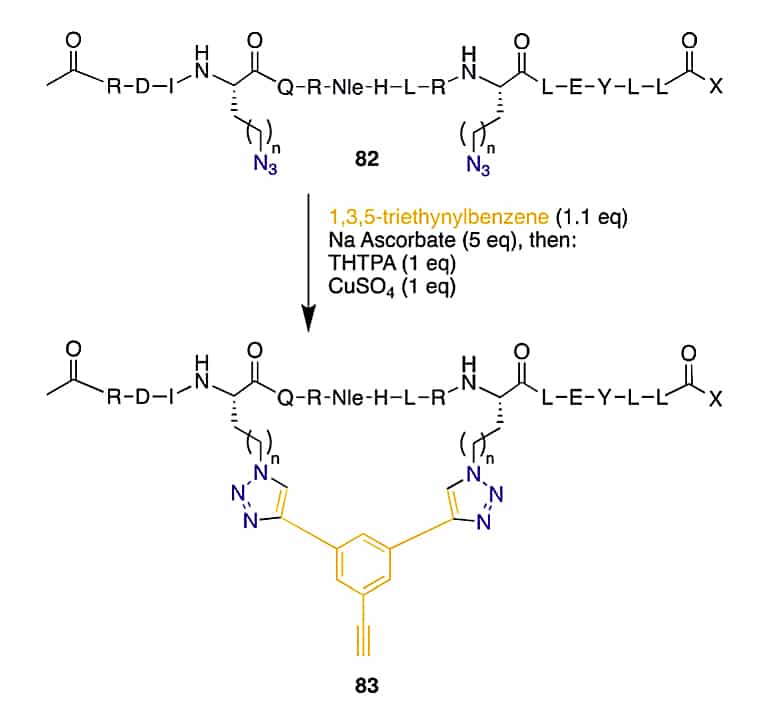
CuAAC can perform in either on-resin or solution without protecting groups under mild conditions. Once introduce two ω-azido amino acids within a peptide, the macrocyclization can be achieved by two CuAACs with a bis-alkynyl linker.
1,5-Disubstituted Triazole Formation(RuAAC)
The 1,5-Disubstituted 1,2,3-triazole has similar H-bond acceptor and donor position as the E-amide bond, it also has function as a bioisostere for E-amide bond. In addition, because the Cα–Cα distance in 1,5-substituted triazole is similar to that in cysteine disulfide, it is also a disulfide mimetic.
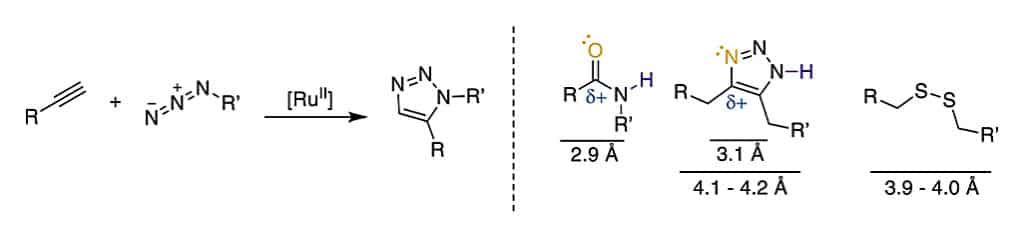
The 1,5-disubstituted counterparts is regioselectively by the Ru catalyzed cycloaddition. Comparing to the CuAAC, the internal alkynes also can undergo RuAAC with introduction of additional substituents.
Multiple researches indicate the higher bioisostery of 1,5-sbstituted triazoles over 1,4-substituted triazoles. Further researches underline the 1,5-disubstutited triazoles as disulfide isosteres can improve pharmacokinetic and change pharmacokinetic.
Metal-free 1,5-Disubstituted Triazole
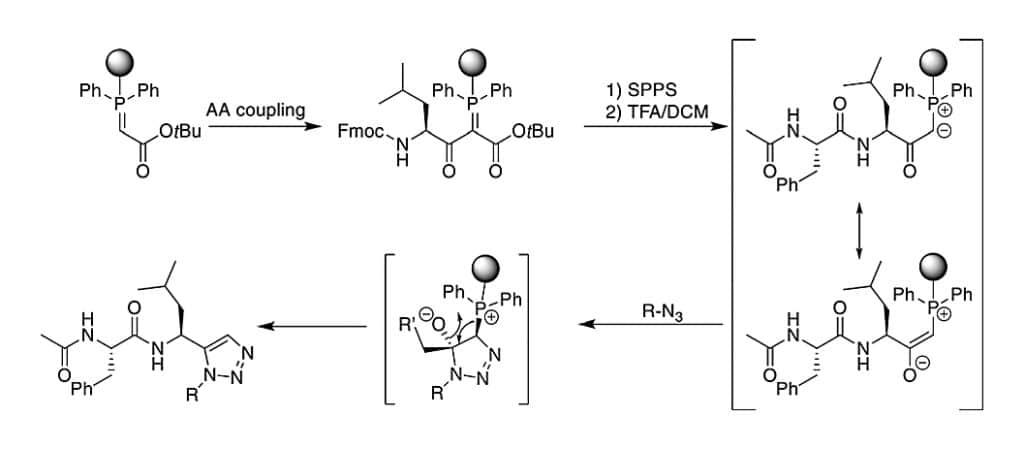
There is a metal-free synthesis method of 1,5-disubstituted triazole formation.
- The resin-bound phophsphoranylidene acetate reacts with the carboxyl group of the Fmoc-amino acid.
- Then apply the standard SPPS to elongate the amino acids.
- Apply acid treatment to liberate α-carbonyl phosphorous ylide.
- After treatment with the azide, create 1,5-disubstituted triazole directly.
This is transferred subsequently to the on-resin cyclizaion of peptides.
Multicomponent Macrocyclization
As the increased interest in peptide macrocyclization, there are different multicomponent reactions.
Ugi Reaction
The Ugi reaction is a 4-component reaction, it combines isocyanide, amino, carboxylic acid, and aldehyde functionalities to create the N-methylated amides. Ugi reaction can increase the molecular diversity in ring closing step by the application of non-amino-acid building blocks.
The Ugi reaction is applied in the synthesis of head-to-tail cyclic peptides, form liner peptides and conventional aldehydes. With the amino aldehyde, the attack of exocyclic nucleophilic aziridine is very fast. The non-nucleophilic solvent can prevent the premature solvolysis of the anhydirde.
The activated aziridine ring within the cyclic peptide can undergo nucleophilic ring opening for later bio-conjugation.
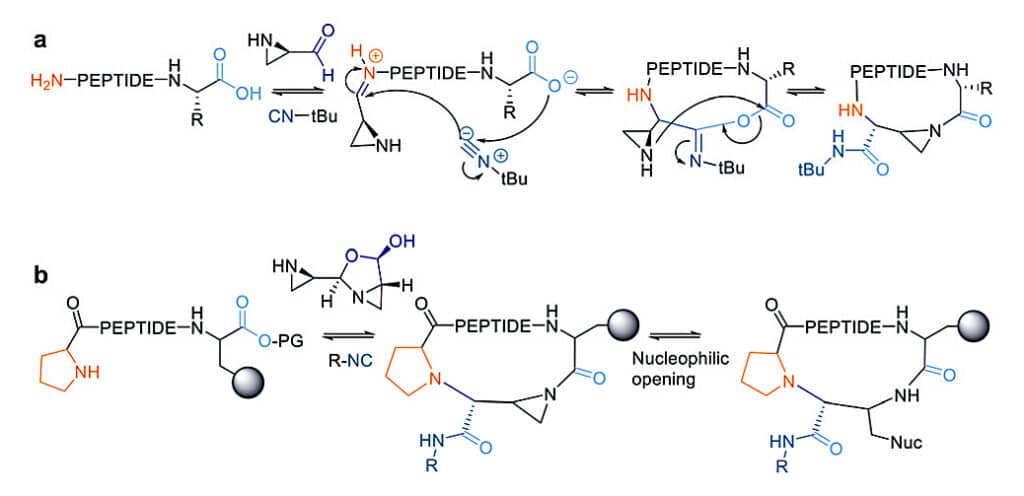
The Ugi reaction is also suitable for other cyclization, like sidechain to sidechain, sidechain to head, sidechain to tail. These cyclic reactions are faster and more efficient than head-to-tail, since the higher flexibility of the sidechains. By careful selection of the isocyanide component, the diversity of peptide scaffold are increase by the N-substitution of newly formed amide. This method is employed to stabilize secondary structures and functionalize sidechain-tethering lactam simultaneously.
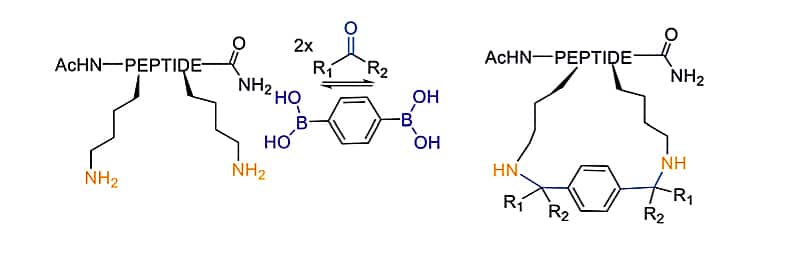
As for cyclization chemistry, the scaffold strategy is used for Ugi multi component reactions (MCR). The liner peptides with two acidic amino acids can couple with diisocyanide scaffolds, in order to generate sidechain-to-sidechain cyclic peptides.
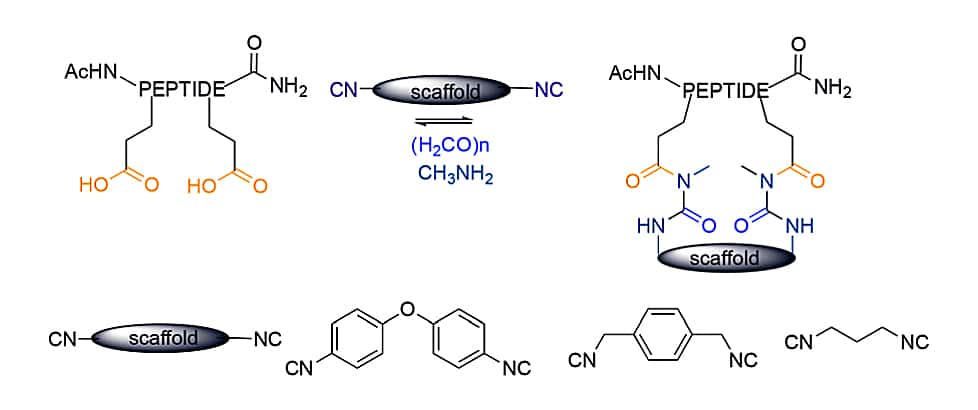
The macrocyclization via Ugi MCR can combine with subsequent disulfide formation to generate bicyclic peptides. This sulfur-switchUgi reaction synthesize disulfide-linked cyclic peptides from four components, then oxidative cycliztion of two cysteines with I2 to form disulfides.
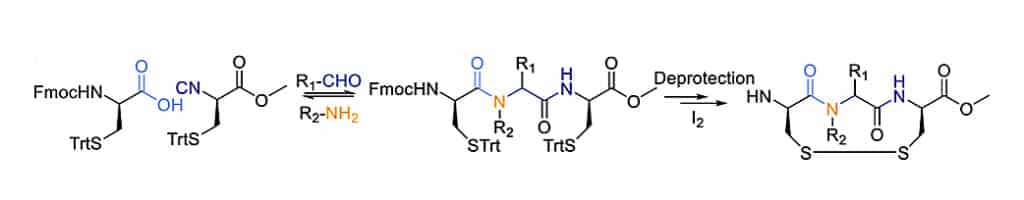
In order to generate macrocyclic peptides in solution and on-resin, the carboxylic acid in the classic four-component can be replaced by an electron-poor phenol (3-nitrotyrosine), and yield tertiary nitroanilines in the Ugi-smiles reaction.
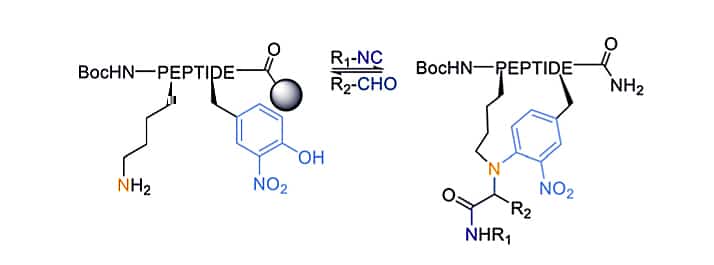
Petasis Reaction
Petasis reaction is also known as Borono-Mannich reaction. It is applied for peptide late-stage diversification on-resin and peptide stapling in solution. This three-component condensation depends on the imine formation via transamination.
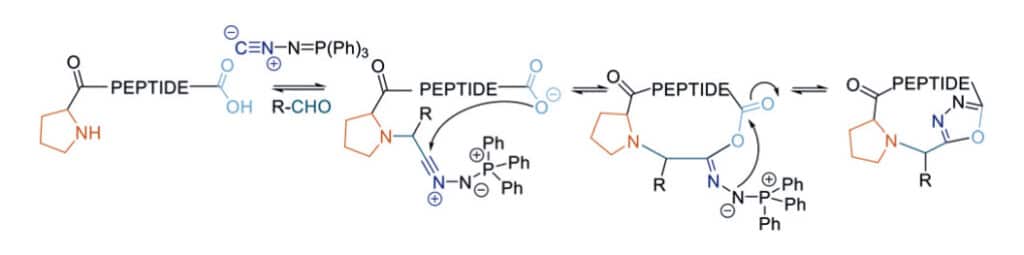
1,3,4-Oxadiazole Formation
The head-to-tail peptidomimetic is synthesized in one step multicomponent reaction of an aldehyde, linear peptide, and (N-isocyanimino)triphenylphosphorane. The final backbone contains the 1,3,4-oxadiazole formation. In contract to the analogous homodetic macrocycles, it can stabilize the unique intramolecular hydrogen-bond network and enable the high passive membrane permeability.
Conclusion
The favorable properties peptidic macrocycles and peptidomimetics led to the rapid evolution of peptide chemistry. As more and more mild, specific, and mutually orthogonal cycilization method, chemically diverse peptides and peptidomimetics have high possibility for synthesizing in the controlled manner.
Qyaobio has more than 15 years experience in synthesis of cyclic peptides, contact us now for your novel peptide synthesis.